SPARKLE
SPARKLE - Single-cell Phenotype Association Research Kit for Large-scale dataset Exploration 
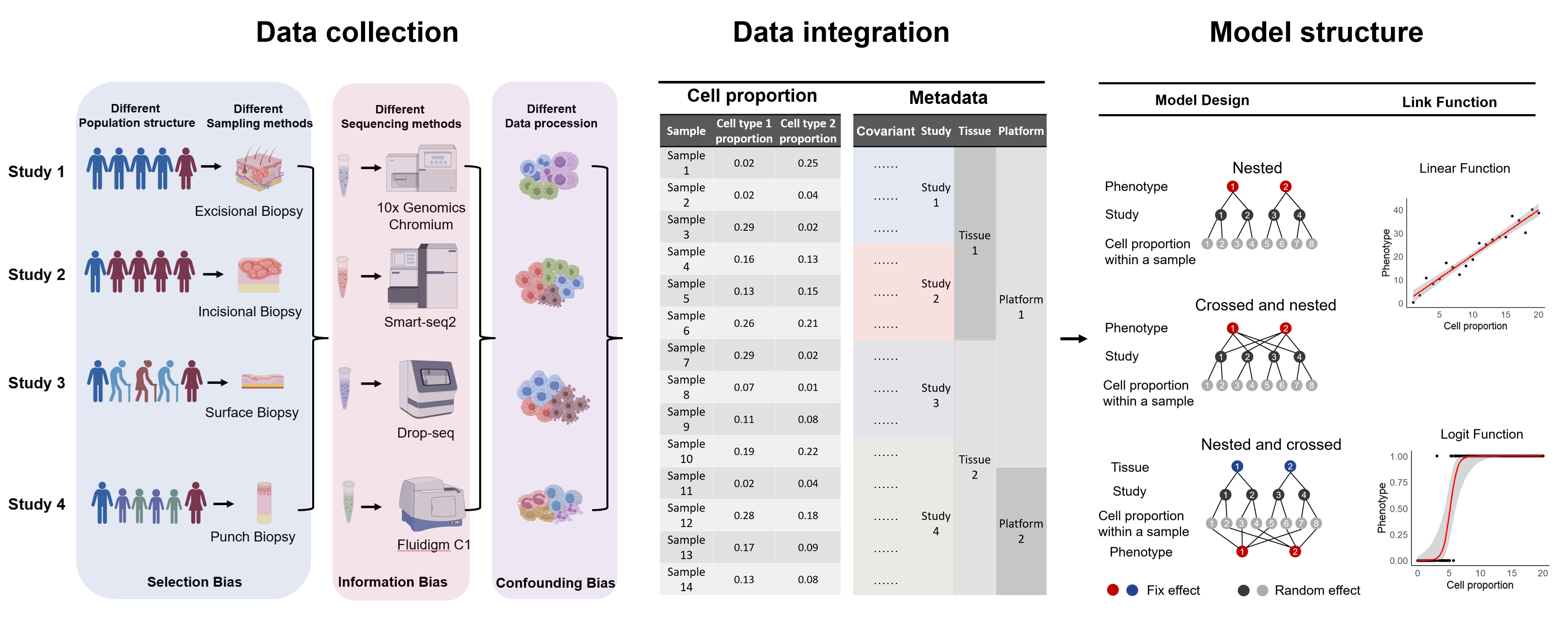
SPARKLE is based on generalized linear mixed models (GLMM) for large-scale single-cell cell-phenotype association analysis. SPARKLE supports the flexibly inclusion of metadata variables as covariates to mitigate the impact of heterogeneity on result accuracy.
Installation
Running the package requires a working R environment (>=3.5).
if (!requireNamespace("devtools", quietly = TRUE)) {
install.packages("devtools")
}
devtools::install_github("chenxi199506/SPARKLE", dep=TRUE, force=TRUE)
Warning: For Linux or MacOS users, if cmake is not installed, there might be errors during the installation of lme4 and bruceR dependencies. Please install the following first:
conda install -c anaconda cmake
conda install -c conda-forge r-lme4
conda install -c bioconda r-brucer
Capabilities
-
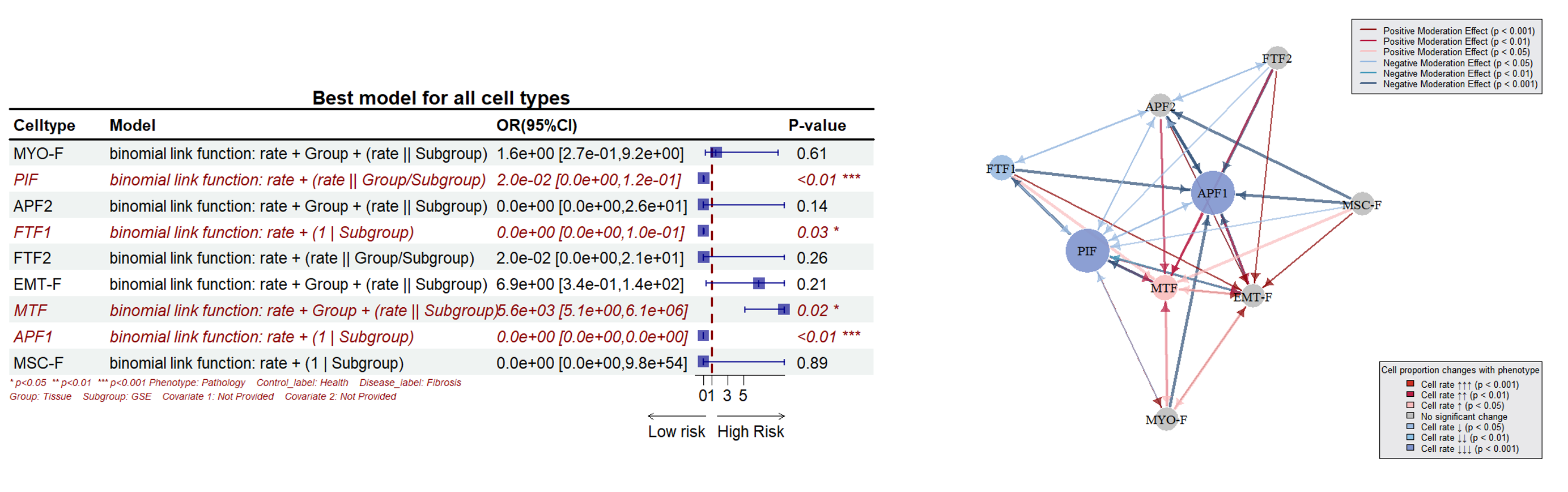
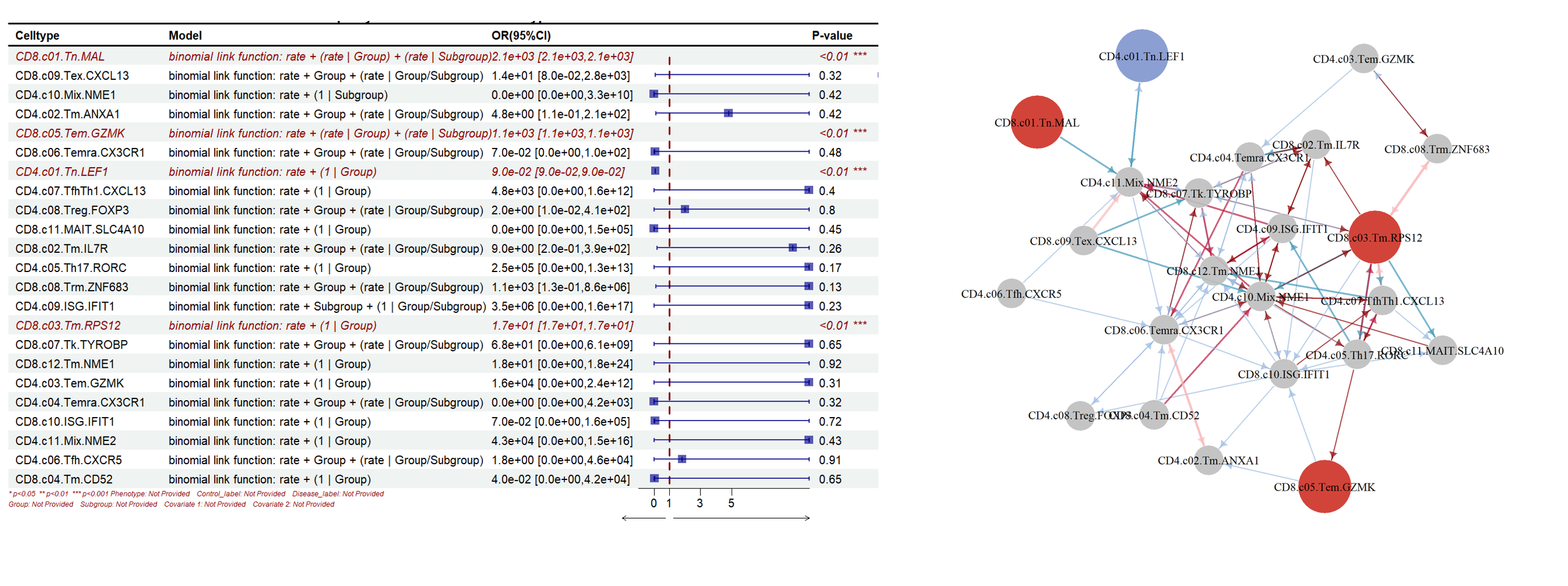
SPARKLE can perform precise statistical tests for different cell proportion-phenotype associations, identify key cell types significantly associated with phenotypes, calculate effect sizes with odds ratios (OR), and generate forest plots.(SPRAKLE 可以对不同的细胞比例-表型关联进行精确的批量统计学检验,寻找跟表型显著相关的关键细胞类型,计算效应值的OR值并绘制森林图)
-
SPARKLE supports confounding analysis, mediation effect analysis, and masking effect analysis for tool cells, explaining how tool cells influence the association between target cells and phenotypes, calculating effect sizes with OR, and generating forest plots.(SPARKLE 支持工具细胞混杂分析、中介效应分析和遮掩效应分析,解释工具细胞如何影响了目标细胞和表型的关联,计算效应值的OR值并绘制森林图)
-
SPARKLE supports moderation effect analysis between cells and generates network diagrams.(SPARKLE支持细胞间的调节效应分析,绘制网络图。)
Tutorials
SPARKLE provides 4 cases for users. All data involves in the cases were in the package.
Tutorials 01: Supported Data Input Formats
Tutorials 02: Example Case: COVID19 dataset
Tutorials 03: Example Case: MNP dataset